
Sponsors should review the guidance Formal Meetings with Sponsors and Applicants for PDUFA Products for information on formal meetings with sponsors and applicants. After the IND submission has been delivered to the FDA, it undergoes a review process with several possible outcomes. This page itemizes potential FDA responses and the steps an investigator should take in each situation.
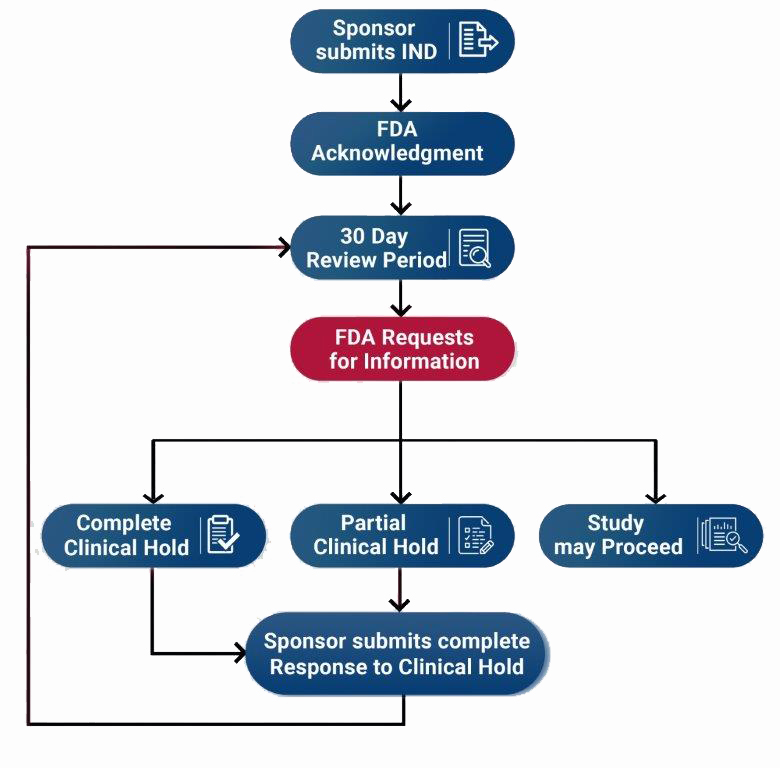
Sponsors should review the guidance Formal Meetings with Sponsors and Applicants for PDUFA Products for information on formal meetings with sponsors and applicants. After the IND submission has been delivered to the FDA, it undergoes a review process with several possible outcomes. This page itemizes potential FDA responses and the steps an investigator should take in each situation.
Prior to the submission of the initial IND, the sponsor may request a formal meeting with FDA-reviewing officials via a pre-IND meeting, as defined in 21 CFR 312.82. The FDA allows for one pre-IND meeting prior to IND submission to discuss any questions or concerns concerning clinical trial approach.
In the process of drug development, a pre-IND meeting is valuable in planning a drug development program, and can provide sponsors information that will assist them in preparing to submit complete investigational new drug applications.
Some situations when a pre-IND meeting could be requested would be when:
- the product is intended to treat a serious or life-threatening disease
- there is a novel indication
- there are no current guidance documents
- there are pharmacologic or toxicology signals of concern
- the drug is a new molecular entity
The primary purpose of this meeting is to review and reach agreement on the design of animal studies needed to initiate human testing. The meeting may also provide an opportunity for discussing the scope and design of phase 1 testing, plans for studying the drug product in pediatric populations, and the best approach for presentation and formatting of data in the IND. During the meeting, the chemistry, manufacturing, and controls (CMC) information, nonclinical plan, and initial clinical plan is presented to the FDA with scientific justification for any non-standard aspect.
In addition to an in-person meeting, a written response can also be requested. The FDA will respond with the meeting date and the type of meeting granted. The study team will then need to send a briefing package 30 days prior to the meeting with finalized questions and information. The FDA will respond to the briefing package about 24 hours before the meeting, providing the sponsor time to review the FDA's responses prior to the meeting. If their written response is sufficient, the meeting can be cancelled, or the meeting should be focused on any outstanding questions remaining. No new questions or proposals may be raised during the pre-IND meeting.

The timing of the pre-IND meeting often depends on the complexity of the discussion topics. Sponsors that prefer to obtain confirmation of toxicology plans and manufacturing will benefit from a meeting held 6 months to 1 year prior to the planned IND submission. In such cases, a meeting request should be submitted approximately 60 days prior to the desired meeting date. The FDA will respond to a request for a pre-IND meeting within 21 days of receiving the request.
Information to Include in a Pre-IND Meeting Request
The process for requesting a pre-IND meeting starts by submitting a formal letter to the responsible project management staff or other designated personnel of the appropriate division within CDER or CBER. In order for the request to be as complete as possible and to give the FDA an accurate understanding of what the sponsor hopes to obtain from the meeting, the meeting request should contain the following elements:
- Product name and application number
- Chemical name and structure
- Brief product description
- Proposed indication or context of product development
- Meeting purpose and objectives including:
- Background information
- Summary of studies that have been completed
- Clinical study plan
- Nature of questions to be asked
- Proposed agenda, including estimated times needed for each agenda item
- Listing of specific questions categorized and grouped by discipline.
- For example: CMC, pharmacology/toxicology, biostatistics, clinical investigations
- List of meeting participants attending on behalf of the sponsor, including titles and affiliations
- List of FDA staff or disciplines requested to participate in the meeting (optional)
- Quantitative composition (all ingredients by percent composition) of the drug proposed for use
- Suggested meeting dates and times
- Format of the meeting (e.g. face-to-face, teleconference, video conference)
Pre-IND Briefing Package
Meeting packages are much more detailed than meeting request letters and should provide summary information relevant to the product, organized according to the proposed agenda. If the intent of the meeting is to discuss clinical requirements of the pre-IND package, the sponsor should submit a proposed clinical trial protocol and statistical analysis plan, to which the FDA will respond after a review. The principal aim of the pre-IND meeting is to ensure that the drug development plan and future clinical trials are going to be acceptable to the FDA. This is an opportunity for sponsors to gain valuable feedback from leaders in the industry. It is important that sponsors remember that maximum transparency with intended clinical plans leads to maximum meeting value.
The pre-IND briefing package must be provided to the FDA at least 30 days prior to the meeting date. The purpose of this package is to provide the FDA with:
- background information on the chemical development concept
- information on the active ingredient
- an initial clinical and preclinical development strategy
- a future development strategy including product scale-up and final formulation, and proposed animal and clinical studies
- a clear and concise overview of the planned development program
Providing the package before the meeting allows FDA the opportunity to comment on a proposed program of development and should include:
- Overall program synopsis
- Whether the animal efficacy rule is being considered
- Clinical study synopsis to obtain FDA input on inclusion, exclusion, and endpoints
- Results for in vitro and early in vivo toxicology
- Rationale for safety, based on toxicological profile and safety margin using dose regimen and exposure
- Brief description of the manufacturing scheme for the active pharmaceutical ingredient (API) and formulation for clinical study
- Brief assay descriptions
- Full description of the development plan
- Copy of the meeting request with updates to reflect the most current information.
Conduct of the Pre-IND Meeting
During the meeting, the presentation should stay focused on the agenda and should not include data that has not been included in the briefing package. It is critical that questions to the FDA are specific and well-phrased so that clear, concise feedback can be provided. The questions should be prioritized relative to importance of the issue, and FDA should be made completely aware of any concerns. Questions for the FDA should be posed in such a way that the agency can either agree or disagree with the question. Example questions may include:
- Does the agency agree that no additional toxicology testing is required to support the proposed clinical study?
- Does the agency agree that the proposed clinical study design, including the primary endpoint, visit schedule, study procedures, and inclusion/exclusion criteria, are acceptable?
- Does the agency agree with the proposed statistical analysis plan?
Recurrent problems have been identified at pre-IND meetings, so the sponsor should aim to avoid providing inadequate or insufficient CMC information and pre-clinical support, unacceptable clinical trial design, non-compliance with GCP, and a lack of information on the dosage selection.
After the Pre-IND Meeting
After approximately 30 days, FDA will provide the final written responses to the questions and note the discussions had during the meeting and the agreements made. Responses to questions are based on the best judgement of FDA relative to current industry practices and knowledge base at the time of the meeting. Feedback provided in the FDA meeting is not “binding” and FDA could change their opinion of what should be done if relevant new information is available before submission of the IND.
The sponsor should review the meeting minutes very carefully and if there is a question that needs further clarification or there is a dispute of a specific FDA response, the FDA project manager should be notified. The question or dispute will be reviewed within the FDA and discussed within the FDA. The sponsor's concerns will be taken under consideration by the review division and the office director if the office director was present at the meeting.
If the minutes are deemed to accurately reflect the meeting discussion, the point of contact will convey this decision to the sponsor or applicant and the minutes will stand as the official documentation of the meeting. After discussions with the sponsor, if the FDA deems it necessary to effect a change to the official minutes, the changes will be documented in an addendum to the official minutes. The addendum will also document any continued sponsor or applicant objections.
Building a strong relationship with the FDA is essential for a successful drug program. This begins with taking advantage of the valuable information that can be gleaned from pre-IND meetings. Proper utilization of pre-IND meetings may reduce the drug's time to market and ensure that the proposed studies are designed to provide useful information. Sponsors who are forthcoming with potential issues of concern during the drug development process will benefit from input provided by the regulatory agency.
In addition to pre-IND meetings, the FDA offers other opportunities to formally engage for guidance on development programs.
Type A Meetings
Type A meetings are held to help move along a stalled development program. Some examples of these may include:
- Dispute resolution meetings
- Meetings to discuss clinical holds
- The sponsor may ask for input from FDA on how to address comments raised in the clinical hold letter, or may discuss with FDA a new path forward if the sponsor's previous clinical hold responses were not deemed adequate for removing the hold.
- Meetings for protocols submitted under procedures of an FDA Nonagreement Special Protocol Assessment
- Post-action meetings within 90 days of the sponsor receiving a regulatory action, non-approval letter from FDA
- Meetings within 30 days of the FDA issuing a refuse-to-file letter
Type B Meetings
Pre-IND meetings fall under the Type B meeting category. In addition, examples include:
- Pre-emergency use meetings
- Post-action meetings after 90 days of the sponsor receiving a regulatory action, non-approval letter from FDA,
- Meetings discussing REMS programs
- Meetings discussing products with breakthrough therapy designation status
Type C Meetings
Type C meetings consist of those that do not fall under the Type A or Type B categories. Type C meetings may be requested by the sponsor to discuss the development and review of a product, like a new biomarker or surrogate endpoint used in the proposed context.